
2026年1月14日,中國醫學科學院系統醫學研究院/蘇州系統醫學研究所(簡稱系統所)李貴登團隊聯合Fred Hutchinson癌癥研究中心的Philip D. Greenberg團隊,在 Nature 《自然》和 Immunity & Inflammation 《免疫與炎癥》上同步發表題為“ The Ubiquitin Ligase KLHL6 Drives Resistance to CD8 + T Cell Dysfunction ”(泛素連接酶KLHL6促進CD8+ T細胞抵抗功能障礙)和“ Chronic TCR Signaling - Driven Suppression of the FOXO1 - KLHL6 Axis Promotes T Cell Exhaustion ”(長期TCR信號通過抑制FOXO1-KLHL6信號軸驅動T細胞耗竭)的研究論文,揭示了持續TCR刺激導致的E3泛素連接酶KLHL6的下調,是驅動T細胞走向耗竭的關鍵分子事件。這一發現不僅回答了“持續TCR信號如何驅動T細胞耗竭”這一關鍵科學問題,還為開發新型T細胞免疫治療策略提供了極具潛力的新靶點,有望推動CAR-T、TCR-T等細胞免疫治療進一步突破當前普遍面臨的“耗竭瓶頸”。
在腫瘤免疫的“戰場”上,CD8? T細胞猶如沖鋒在一線的士兵。它們通過T細胞受體(TCR)識別腫瘤抗原,從而精準定位并清除癌細胞。然而,在腫瘤或慢性感染的環境中,這些“士兵”往往陷入持久戰。持續暴露于抗原信號的刺激下,它們會逐漸進入一種稱為“T細胞耗竭”的狀態,表現為殺傷功能下降、代謝能力減弱,最終難以有效執行抗腫瘤任務。研究顯示,耗竭T細胞并非完全失去功能。其中一部分仍保留了自我更新潛質,能夠響應PD-1抗體等免疫治療,被稱為“前體耗竭T細胞”,可視為仍有戰斗潛力的“預備役”;而另一部分則進入幾乎不可逆轉的“終末耗竭”狀態,很難被重新激活。終末耗竭T細胞的比例升高,是導致腫瘤免疫治療失敗的一個重要原因。
李貴登團隊前期發現甘露糖代謝和酸性微環境能夠有助于維持“干性樣”的前體耗竭T細胞分化。然而,T細胞耗竭的具體發生機制,長期以來一直是T細胞生物學領域一個懸而未決的核心科學問題。腫瘤抗原觸發的TCR信號是啟動T細胞抗腫瘤免疫應答的初始“鑰匙”,但令人費解的是,這種信號的持續、反復激活,最終卻將T細胞推向了功能耗竭的狀態,導致其喪失抗腫瘤能力。這背后究竟隱藏著怎樣的分子調控網絡與關鍵轉化節點?
在這項研究中,李貴登團隊設計了一種基于計算分析指導的“雙重”CRISPR篩選策略,成功鑒定出KLHL6作為能夠同時調控T細胞耗竭程序和細胞內能量代謝的關鍵泛素化酶。后續實驗表明,增強KLHL6表達可顯著提升T細胞的腫瘤殺傷能力并改善其線粒體功能。通過追蹤T細胞的分化路徑,研究進一步確認KLHL6過表達能夠有效阻止Tpex細胞向終末耗竭狀態分化,并通過維持Tpex細胞的自我更新和存活能力,實現持久抗腫瘤免疫應答。
進一步機制解析發現,TOX和PGAM5是KLHL6的直接作用底物。在慢性感染或腫瘤微環境中,持續性TCR信號刺激引起KLHL6表達持續下調,進而削弱對TOX的泛素化降解,促使TOX蛋白積累,加速T細胞耗竭程序的啟動及向終末表型分化。同時,KLHL6表達下調還會通過PGAM5-Drp1信號軸誘導線粒體過度分裂和呼吸功能損傷,進一步加劇T細胞耗竭程度(見圖)。
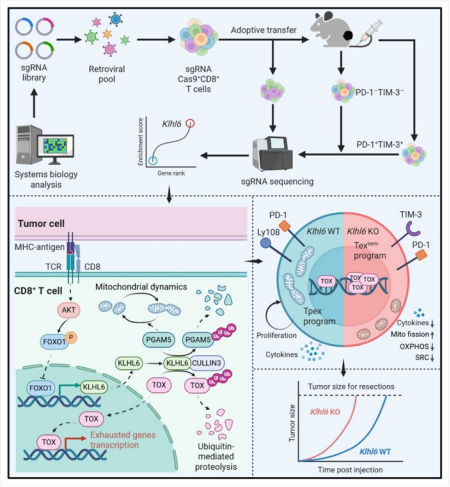
這一基礎生物學發現確立了泛素連接酶KLHL6在腫瘤環境下調控T細胞功能的核心地位,也為理解持續抗原刺激下TCR信號如何從激活信號轉變為耗竭程序提供了機制性解答。該發現為靶向KLHL6的工程化T細胞改造策略奠定了理論基礎,并揭示了潛在的干預靶點,為進一步提升T細胞免疫治療的療效開辟了新的思路。
該研究工作得到中國醫學科學院醫學與健康科技創新工程(2024-I2M-ZD-009)、中國醫學科學院中央級公益性科研院所基本科研業務費(2021-RC310-014)、國家自然科學基金(32525028)等項目的資助。
系統所李貴登研究員與Fred Hutchinson癌癥研究中心Philip D. Greenberg教授為論文的共同通訊作者,系統所副研究員程洪成、Fred Hutchinson癌癥研究中心蘇亞鵬博士及北京協和醫學院博士研究生潘曉麗為該論文的共同第一作者。
論文鏈接:
https://www.nature.com/articles/s41586-025-09926-8
https://link.springer.com/article/10.1007/s44466-025-00023-z
供稿:系統院

